PLOS ONE
Abstract
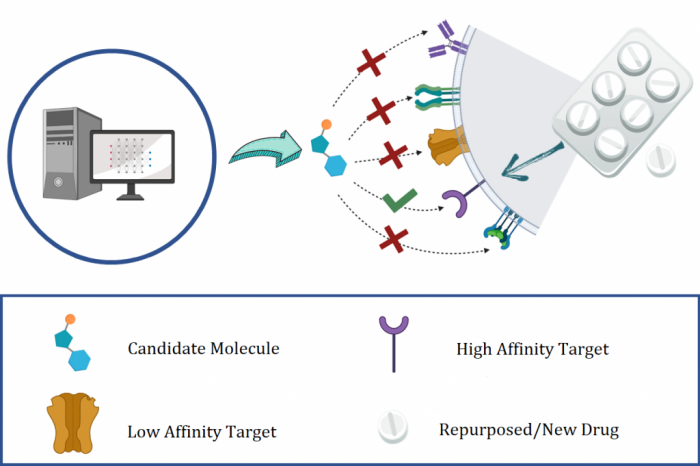
The discovery and development of novel pharmaceuticals is an area of active research mainly due to the large investments required and long payback times. As of 2016, the development of a novel drug candidate required up to $ USD 2.6 billion in investment for only 10% rate of approval by the FDA. To help decreasing the costs associated with the process, a number of in silico approaches have been developed with relatively low success due to limited predicting performance. Here, we introduced a machine learning-based algorithm as an alternative for a more accurate search of new pharmacological candidates, which takes advantage of Recurrent Neural Networks (RNN) for active molecule prediction within large databases. Our approach, termed PharmaNet was implemented here to search for ligands against specific cell receptors within 102 targets of the DUD-E database, which contains 22886 active molecules. PharmaNet comprises three main phases. First, a SMILES representation of the molecule is converted into a raw molecular image. Second, a convolutional encoder processes the data to obtain a fingerprint molecular image that is finally analyzed by a Recurrent Neural Network (RNN). This approach enables precise predictions of the molecules’ target on the basis of the feature extraction, the sequence analysis and the relevant information filtered out throughout the process. Molecule Target prediction is a highly unbalanced detection problem and therefore, we propose that an adequate evaluation metric of performance is the area under the Normalized Average Precision (NAP) curve. PharmaNet largely surpasses the previous state-of-the-art method with 97.7% in the Receiver Operating Characteristic curve (ROC-AUC) and 65.5% in the NAP curve. We obtained a perfect performance for human farnesyl pyrophosphate synthase (FPPS), which is a potential target for antimicrobial and anticancer treatments. We decided to test PharmaNet for activity prediction against FPPS by searching in the CHEMBL data set. We obtained three (3) potential inhibitors that were further validated through both molecular docking and in silico toxicity prediction. Most importantly, one of this candidates, CHEMBL2007613, was predicted as a potential antiviral due to its involvement on the PCDH17 pathway, which has been reported to be related to viral infections.